Pratip Chakraborty, Rafael C. Couto, and Nanna H. List
J. Phys. Chem. A 2023, 127, 25, 5360–5373
Abstract
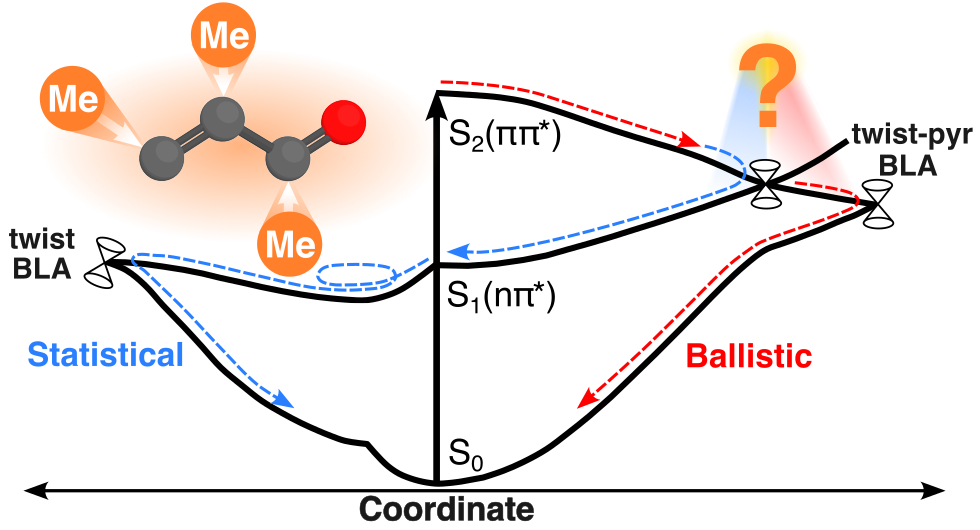
Chemical substituents can influence photodynamics by altering the location of critical points and the topography of the potential energy surfaces (electronic effect) and by selectively modifying the inertia of specific nuclear modes (inertial effects). Using nonadiabatic dynamics simulations, we investigate the impact of methylation on S2(ππ) internal conversion in acrolein, the simplest linear α,β-unsaturated carbonyl. Consistent with time constants reported in a previous time-resolved photoelectron spectroscopy study, S2 → S1 deactivation occurs on an ultrafast time scale (∼50 fs). However, our simulations do not corroborate the sequential decay model used to fit the experiment. Instead, upon reaching the S1 state, the wavepacket bifurcates: a portion undergoes ballistic S1 → S0 deactivation (∼90 fs) mediated by fast bond-length alternation motion, while the remaining decays on the picosecond time scale. Our analysis reveals that methyl substitution, generally assumed to mainly exert inertial influence, is also manifested in important electronic effects due to its weak electron-donating ability. While methylation at the β C atom gives rise to effects principally of an inertial nature, such as retarding the twisting motion of the terminal −CHCH3 group and increasing its coupling with pyramidalization, methylation at the α or carbonyl C atom modifies the potential energy surfaces in a way that also contributes to altering the late S1-decay behavior. Specifically, our results suggest that the observed slowing of the picosecond component upon α-methylation is a consequence of a tighter surface and reduced amplitude along the central pyramidalization, effectively restricting the access to the S1/S0-intersection seam. Our work offers new insight into the S2(ππ) internal conversion mechanisms in acrolein and its methylated derivatives and highlights site-selective methylation as a tuning knob to manipulate photochemical reactions.