Edward G. Hohenstein, Jimmy K. Yu, Christoph Bannwarth, Nanna Holmgaard List, Alexander C. Paul, Sarai D. Folkestad, Henrik Koch, Todd J. Martínez
J. Chem. Theory Comput. 2021, 17, 7120
Abstract
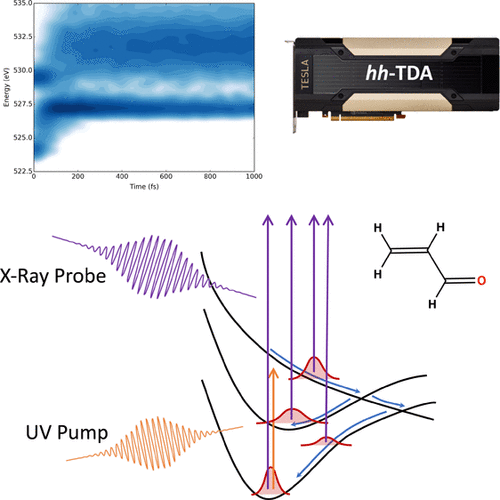
Time-resolved near-edge X-ray absorption fine structure (TR-NEXAFS) spectroscopy is a powerful technique for studying photochemical reaction dynamics with femtosecond time resolution. In order to avoid ambiguity in TR-NEXAFS spectra from nonadiabatic dynamics simulations, core- and valence-excited states must be evaluated on equal footing and those valence states must also define the potential energy surfaces used in the nonadiabatic dynamics simulation. In this work, we demonstrate that hole–hole Tamm–Dancoff-approximated density functional theory (hh-TDA) is capable of directly simulating TR-NEXAFS spectroscopies. We apply hh-TDA to the excited-state dynamics of acrolein. We identify two pre-edge features in the oxygen K-edge TR-NEXAFS spectrum associated with the S2 (ππ*) and S1 (nπ*) excited states. We show that these features can be used to follow the internal conversion dynamics between the lowest three electronic states of acrolein. Due to the low, O(N2) apparent computational complexity of hh-TDA and our GPU-accelerated implementation, this method is promising for the simulation of pre-edge features in TR-NEXAFS spectra of large molecules and molecules in the condensed phase.