C. M. Jones, N. H. List, T. J. Martínez
Abstract
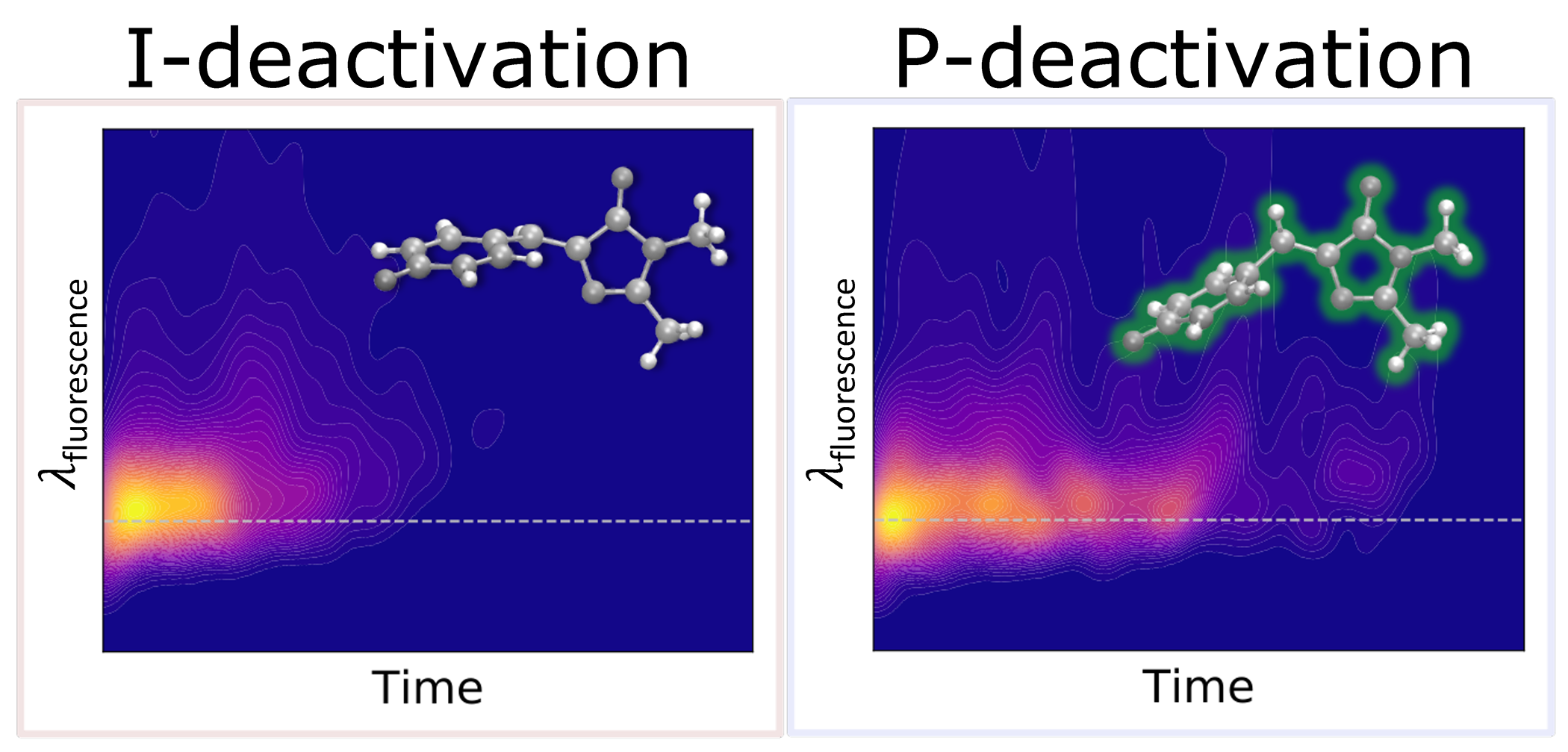
The chromophore of the green fluorescent protein (GFP) is critical for probing environmental influences on fluorescent protein behavior. Using the aqueous system as a bridge between the unconfined vacuum system and a constricting protein scaffold, we investigate the steric and electronic effects of the environment on the photodynamical behavior of the chromophore. Specifically, we apply ab initio multiple spawning to simulate five picoseconds of nonadiabatic dynamics after photoexcitation, resolving the excited-state pathways responsible for internal conversion in the aqueous chromophore. We identify an ultrafast pathway that proceeds through a short-lived (sub-picosecond) imidazolinone-twisted (I-twisted) species and a slower (several picoseconds) channel that proceeds through a long-lived phenolate-twisted (P-twisted) intermediate. The molecule navigates the non-equilibrium energy landscape via an aborted hula-twist-like motion toward the one-bond-flip dominated conical intersection seams, as opposed to following the pure one-bond-flip paths proposed by the excited-state equilibrium picture. We interpret our simulations in the context of time-resolved fluorescence experiments, which use short- and long-time components to describe the fluorescence decay of the aqueous GFP chromophore. Our results suggest that the longer time component is caused by an energetically uphill approach to the P-twisted intersection seam rather than an excited-state barrier to reach the twisted intramolecular charge-transfer species. Irrespective of the location of the nonadiabatic population events, the twisted intersection seams are inefficient at facilitating isomerization in aqueous solution. The disordered and homogeneous nature of the aqueous solvent environment facilitates non-selective stabilization with respect to I- and P-twisted species, offering an important foundation for understanding the consequences of selective stabilization in heterogeneous and rigid protein environments.