Chey M. Jones, Nanna H. List, Todd J. Martinez
J. Am. Chem. Soc. 2022, 144, 28, 12732
Abstract
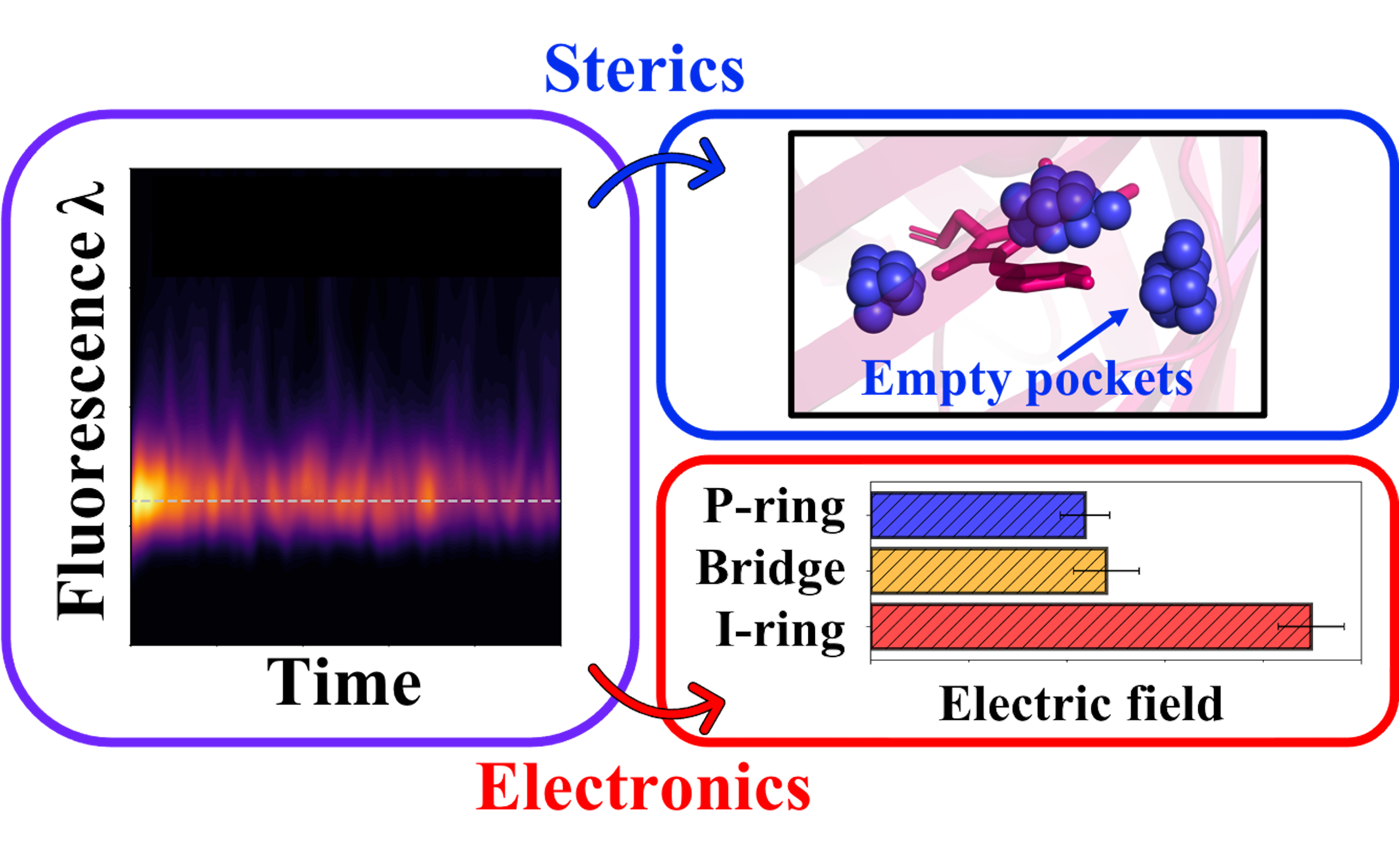
Fluorescent proteins have become routine tools for biological imaging. Yet, their nanosecond lifetimes on the excited state present computational hurdles to a full understanding of these photoactive proteins. In this work, we simulate approximately 0.5 nanoseconds of ab initio molecular dynamics to elucidate steric and electronic features responsible for fluorescent protein behavior. Using green fluorescent protein (GFP) and Dronpa2 – widely used fluorescent proteins with contrasting functionality – as case studies, we leverage previous findings in gas-phase and solution to explore the deactivation mechanisms available to these proteins. Starting with ground-state analyses, we identify steric (the distribution of empty pockets near the chromophore) and electronic (electric fields exerted on chromophore moieties) factors that offer potential avenues for rational design. Picosecond timescale simulations on the excited state reveal that the chromophore can access twisted structures in Dronpa2, while the chromophore is largely confined to planarity in GFP. We couple ab initio multiple spawning (AIMS) and enhanced sampling simulations to discover and characterize conical intersection seams that facilitate internal conversion, which is a rare event in both systems. Our AIMS simulations correctly capture the relative fluorescence profiles of GFP and Dronpa2 within the first few picoseconds, and we attribute the diminished fluorescence intensity of Dronpa2, relative to GFP, to flexible chromophore intermediates on the excited state. Furthermore, we predict that twisted chromophore intermediates produce red-shifted intensities in the Dronpa2 fluorescence spectrum. If confirmed experimentally, this spectroscopic signature would provide valuable insight when screening and developing novel fluorescent proteins.